

limma差异表达分析
本篇笔记的内容是在R语言中利用limma包进行差异表达分析,主要针对转录组测序得到的基因表达数据进行下游分析,并将分析结果可视化,绘制火山图和热图
[TOC]
基因表达差异分析是我们做转录组最关键根本的一步,不管哪种差异分析,其本质都是广义线性模型,limma也是广义线性模型的一种,其对每个gene的表达量拟合一个线性方程。
limma包是2015年发表在Nucleic Acids Resarch一个做差异分析的工具,目前引用次数高达七千多次,最流行的差异分析软件之一就是limma。
环境部署与安装
- 安装limma包
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("limma")
- 安装ggVolcano包
# install.packages("devtools")
devtools::install_github("BioSenior/ggVolcano")
- 安装ggplot2包
#直接安装tidyverse,一劳永逸(推荐,数据分析大礼包)
install.packages("tidyverse")
#直接安装ggplot2
install.packages("ggplot2")
#从Github上安装最新的版本,先安装devtools(如果没安装的话)
devtools::install_github("tidyverse/ggplot2")
输入数据准备
-
样本信息表:第一列为样本名称ID,第二列为样本的分组(CK、HT),
sampleinfo.csv格式如下这一步需要注意:每个分组下至少有两个生物学重复,即至少4行数据,如果没有重复的话在后续贝叶斯检验会出错,原因是该模型利用统计假设检验,多个重复能够评估变异性,而如果仅有一组数据,则无法检验。
sample group A01 CK A02 HT … … -
表达矩阵:每行是一个基因,每列是一个样本,表达数据为TPM,
data.csv格式如下A01 A02 … gene1 xx xx … gene2 xx xx … … … … …
差异表达分析过程
准备环节
载入R包,设置参数,其中job变量用于项目输出文件前缀标识,可以自定义修改。
setwd("D:/LABdata")
options(stringsAsFactors = F)
rm(list=ls()) #清空变量
job <- "test" #设定项目名称
library(limma)
library(ggplot2) #用于绘制火山图
library(ggVolcano)
数据导入
导入样本信息和表达量数据,然后进行删除表达量之和为0基因、log2化、替换异常值等步骤,得到原始数据矩阵。
# 输入表达矩阵和分组文件 -------------------------------------------------------------
expr_data<-read.csv("data.csv",header = T,row.names = 1,sep = ",") #输入文件TPM原始值,行名是基因,列名是样本
expr_data <- expr_data[which(rowSums(expr_data)!=0),] #删除表达量为0的基因
expr_data = log2(expr_data) #log化处理
expr_data[expr_data == -Inf] = 0 #将log化后的负无穷值替换为0
group<-read.csv("sampleinfo.csv",header = T,row.names = 1,sep = ",") #输入文件,样本信息表,包含分组信息
构建分组矩阵
根据样本的分组信息,构建分组矩阵,最终得到的design矩阵由0和1构成,为斜对角矩阵。
# 构建分组矩阵--design ---------------------------------------------------------
design <- model.matrix(~0+factor(group$group))
colnames(design) <- levels(factor(group$group))
rownames(design) <- colnames(expr_data)
构建比较矩阵
设置样本的比较方式,这里为CK对照比HT处理,该步骤生成的文件为1和-1构成的矩阵。
# #构建比较矩阵——contrast -------------------------------------------------------
contrast.matrix <- makeContrasts(CK-HT,levels = design) #根据实际的样本分组修改,这里对照组CK,处理组HT
线性混合模拟
该步骤是limma包的核心步骤,首先使用lmFit函数进行非线性最小二乘法分析,然后用经验贝叶斯调整t-test中方差部分,得到差异表达结果。
# #线性拟合模型构建 ---------------------------------------------------------------
fit <- lmFit(expr_data,design) #非线性最小二乘法
fit2 <- contrasts.fit(fit, contrast.matrix)
fit2 <- eBayes(fit2)#用经验贝叶斯调整t-test中方差的部分
DEG <- topTable(fit2, coef = 1,n = Inf,sort.by="logFC")
DEG <- na.omit(DEG)
最终生成的DEG文件包含以下几列信息:
> colnames(DEG)
[1] "logFC" "AveExpr" "t" "P.Value" "adj.P.Val" "B"
[7] "regulate" "Genes"
差异基因标注
本步骤中对P.Value和logFC进行筛选,并对每个基因进行标注,显示其表达变化情况。
DEG$regulate <- ifelse(DEG$P.Value > 0.05, "unchanged",
ifelse(DEG$logFC > 1, "up-regulated",
ifelse(DEG$logFC < -1, "down-regulated", "unchanged")))
结果保存
生成两个文件,保存差异表达结果。
write.table(table(DEG$regulate),file = paste0(job,"_","DEG_result_1_005.txt"),
sep = "\t",quote = F,row.names = T,col.names = T)
write.table(data.frame(gene_symbol=rownames(DEG),DEG),file = paste0(job,"_","DEG_result.txt"),
sep = "\t",quote = F,row.names = F,col.names = T)
区分上下调基因
该步骤中DEG$P.Value<0.05&abs(DEG$logFC)>1为参数,筛选了差异倍数和显著性,并将基因按照上调和下调进行区分。
DE_1_0.05 <- DEG[DEG$P.Value<0.05&abs(DEG$logFC)>1,]
upGene_1_0.05 <- DE_1_0.05[DE_1_0.05$regulate == "up-regulated",]
downGene_1_0.05 <- DE_1_0.05[DE_1_0.05$regulate == "down-regulated",]
write.csv(upGene_1_0.05,paste0(job,"_","upGene_1_005.csv"))
write.csv(downGene_1_0.05,paste0(job,"_","downGene_1_005.csv"))
差异基因名称提取
提取差异倍数最大的1000个基因,按照顺序保存到txt文本中,只保留基因名称ID,用于后续验证。
tem1 <- head(rownames(upGene_1_0.05),1000) #以logFC差异倍数从大到小为序,提取前1000个基因名称
tem2 <- as.data.frame(tem1) # 转化数据类型为数据框
write.table(tem2$tem,file = paste0(job,"_","upgene_head100_name.txt"),
row.names = FALSE,col.names = FALSE)
tem1 <- head(rownames(downGene_1_0.05),1000) #以logFC差异倍数从大到小为序,提取前1000个基因名称
tem2 <- as.data.frame(tem1) # 转化数据类型为数据框
write.table(tem2$tem,file = paste0(job,"_","downgene_head100_name.txt"),
row.names = FALSE,col.names = FALSE)
自定义筛选阈值
之前的结果均为默认设置,如果你需要修改,仅需更改下面开头两行参数即可,运行后可以得到3个文件,分别是差异基因集、上下调过滤所得基因信息。
foldChange = 2 # 自定义修改筛选参数
padj = 0.05 # 自定义修改筛选参数
All_diffSig <- DEG[(DEG$adj.P.Val < padj & (DEG$logFC > foldChange | DEG$logFC < (-foldChange))),]
#dim(All_diffSig)
write.csv(All_diffSig, paste0(job,"_","all_diffsig_filtered.csv")) ##输出差异基因数据集
### 自定义筛选上调和下调的基因 ===================================================================
diffup <- All_diffSig[(All_diffSig$P.Value < padj & (All_diffSig$logFC > foldChange)),]
write.csv(diffup, paste0(job,"_","diffup_filtered.csv"))
diffdown <- All_diffSig[(All_diffSig$P.Value < padj & (All_diffSig$logFC < -foldChange)),]
write.csv(diffdown, paste0(job,"_","diffdown_filtered.csv"))
利用limma分析后,正常情况下应该会生成下面这些数据文件,可以用于验证过程中是否有问题。
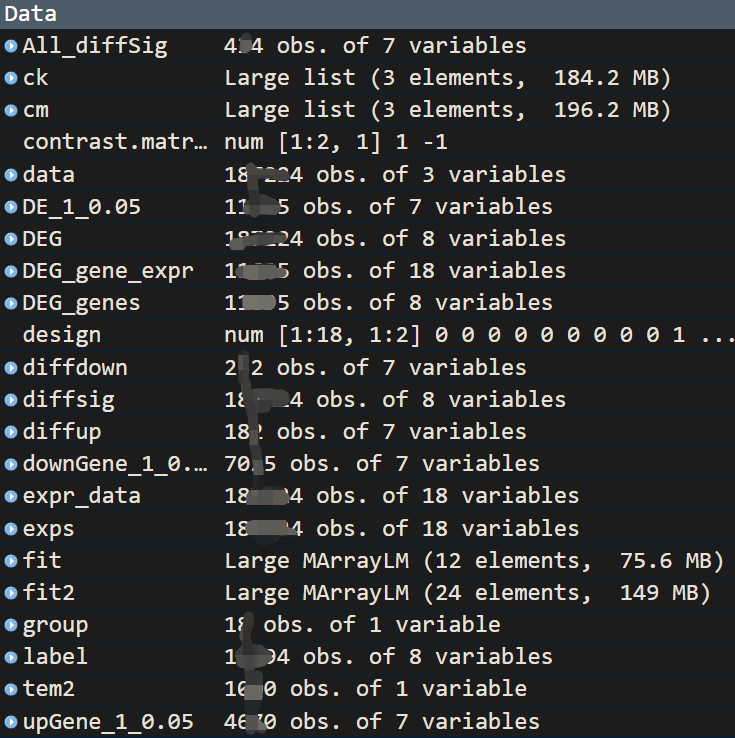
结果可视化
火山图绘制方法一
有了上面的DEG差异分析结果,根据gene、logFC、adj.P.Val三列信息即可绘制火山图,第一种方法使用ggvolcano,绘图代码如下:
# ggvolcano绘制火山图 ----------------------------------------------------------
pdf(paste0(job,"_","volcano1.pdf"),width = 10,height = 10)
ggvolcano(data = DEG,x = "logFC",y = "P.Value",output = FALSE,label = "Genes",
fills = c("#00AFBB", "#999999", "#FC4E07"),
colors = c("#00AFBB", "#999999", "#FC4E07"),
x_lab = "log2FC",
y_lab = "-Log10P.Value",
legend_position = "UR") #标签位置为up right
dev.off()
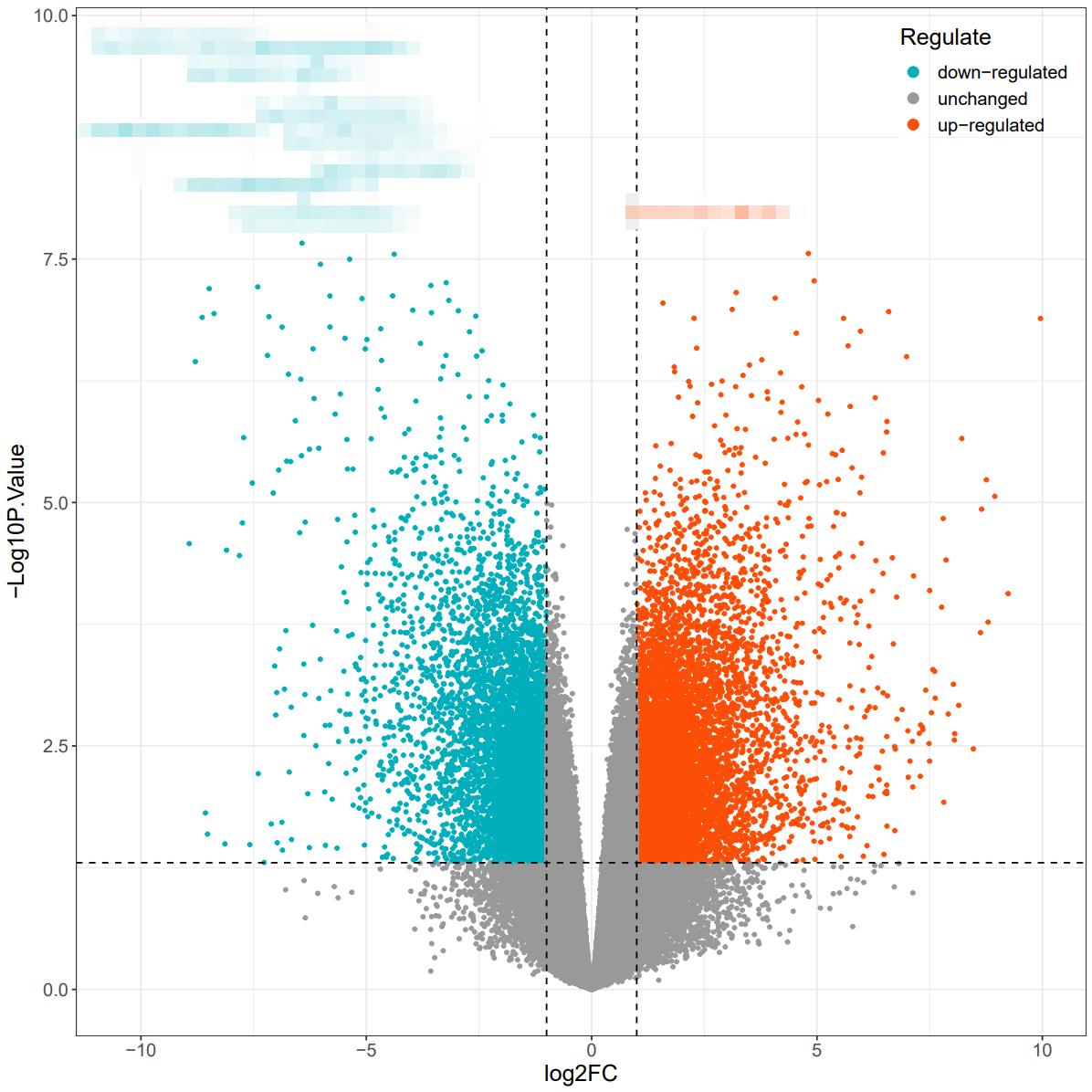
火山图绘制方法二
第二种方法使用ggplot2,得到另外一种形式的火山图,绘图代码如下:
# 火山图的绘制 ------------------------------------------------------------------
DEG$Genes <- rownames(DEG)
pdf(paste0(job,"_","volcano2.pdf"),width = 7,height = 7)
ggplot(DEG,aes(x=logFC,y=-log10(P.Value)))+ #x轴logFC,y轴adj.p.value
geom_point(alpha=0.5,size=2,aes(color=regulate))+ #点的透明度,大小
ylab("-log10(P.Value)")+ #y轴的说明
scale_color_manual(values = c("blue", "grey", "red"))+ #点的颜色
geom_vline(xintercept = c(-1,1),lty=4,col ="black",lwd=0.8)+ #logFC分界线
geom_hline(yintercept=-log10(0.05),lty=4,col = "black",lwd=0.8)+ #adj.p.val分界线
theme_bw() #火山图绘制
dev.off()
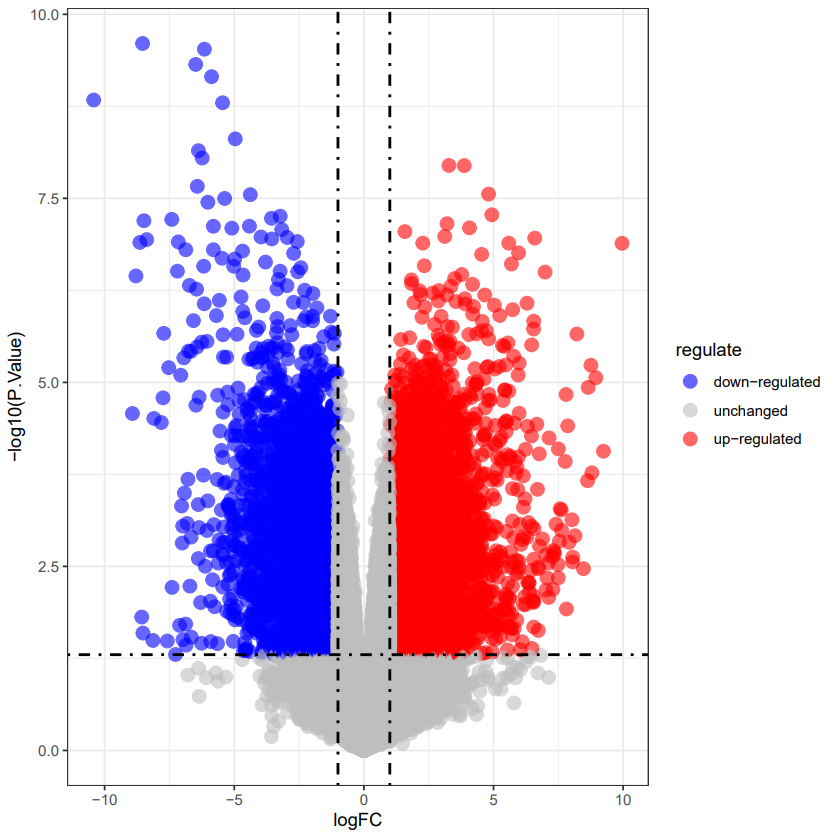
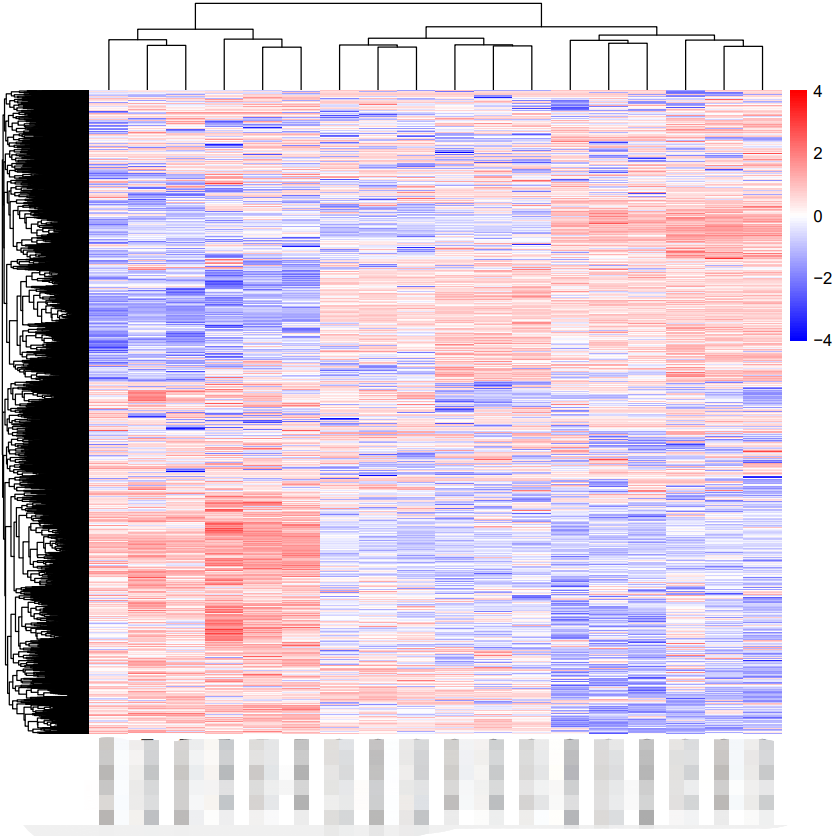
热图绘制
根据基因的表达变化信息,绘制热图并展示聚类树,详细代码如下:
# 热图的绘制 -------------------------------------------------------------------
DEG_genes <- DEG[DEG$P.Value<0.05&abs(DEG$logFC)>1,]
DEG_gene_expr <- expr_data[rownames(DEG_genes),]
#DEG_gene_expr[is.infinite(DEG_gene_expr)] = 0
#DEG_gene_expr[DEG_gene_expr == -Inf] = 0
pdf(paste0(job,"_","pheatmap.pdf"))
pheatmap(DEG_gene_expr,
color = colorRampPalette(c("blue","white","red"))(100), #颜色
scale = "row", #归一化的方式
border_color = NA, #线的颜色
fontsize = 10, #文字大小
show_rownames = F)
dev.off()
参考资料:https://zhuanlan.zhihu.com/p/437712423
本文由mdnice多平台发布
