

原文:How We Used WebAssembly To Speed Up Our Web App By 20X (Case Study)
作者:www.smashingmagazine.com/contact/
译文:如何使用 WebAssembly 来提高我们的 Web 应用的性能 20 倍(实例学习)
译者:Zavier Tang
概述:在这篇文章中,我们将探索如何用编译后的 WebAssembly 替代缓慢的 JavaScript 程序,从而提升 Web App 应用程序的速度。
如果你还没有听说过 WebAssembly,这是它的介绍:WebAssembly 是一种与 JavaScript 一起运行在浏览器中的一种新的语言。没错!也就是说 JavaScript 不再是唯一能在浏览器中运行的语言了!
但是,除了它与 JavaScript 的名称不同之外,它的独特之处在于,你可以从 C / C++ / Rust 等更多的语言编译成 WebAssembly,并在浏览器中运行它们。因为 WebAssembly 是静态类型的,使用线性内存,并以较小的二进制格式存储,所以它的运行速度非常快,可能接近本机原生程序的速度(即以接近于在命令行上运行二进制代码的速度运行)。在浏览器中利用现有的工具和库,以及相关的提速潜力,是 WebAssembly 如此强大的两个原因。
到目前为止,WebAssembly 已被用于各种应用程序,从游戏(如 毁灭战士 3)到将桌面应用程序移植到 Web (如 Autocad 和 Figma。它甚至可以在浏览器之外使用,例如可以作为一种用于 serverless computing 的高效且灵活的语言。
本文是一个使用 WebAssembly 加速数据分析 Web 工具的案例。为此,我们将使用一个用 C 语言编写的可以执行相同计算的程序,将其编译为 WebAssembly,使用它来替换缓慢的 JavaScript。
注意:本文深入研究了一些高级内容,比如编译 C 代码。但是如果你没有这方面的经验,请不要担心,你仍然能够一起来了解 WebAssembly 的功能。
背景介绍
我们将使用的 Web 应用程序是 fastq.bio,它是一个交互式网络工具,为科学家提供快速预览 DNA 测序数据的质量。测序是我们读取 DNA 样本中的 “字母” (即核苷酸)的过程。
下面是应用程序的截图(查看大图):
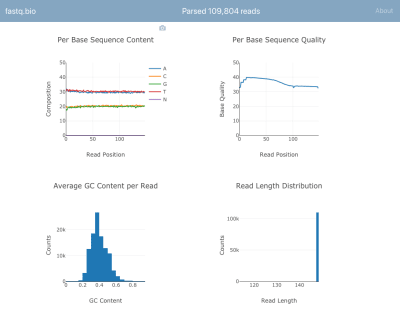
我们不会详细地讨论计算的细节,但是简单地说,上图是为科学家提供了排序进行过程的可视化交换体验,并被用于快速识别数据质量。
尽管有许多的命令行工具可以生成这样的质量控制报告,但是 fastq 的目标是,在不离开浏览器的情况下提供数据质量的交互式预览。对于不熟悉命令行的科学家来说,这非常有用。
应用程序的输入是一个纯文本文件,由测序仪输出,包含 DNA 序列列表和 DNA 序列中每个核苷酸的质量分数。该文件的格式称为 “FASTQ”,因此这个工具被叫做 fastq.bio。
如果您对 “FASTQ” 格式感兴趣,请查看 Wikipedia page 了解更多。
用 JavaScript 实现 Fastq.Bio
在 fastq.bio 的原始版本中,用户首先从计算机中选择一个 FASTQ 文件。使用 File 对象,应用程序从随机的字节位置开始读取一小块数据(使用 FileReader API)。在该数据块中,我们使用 JavaScript 执行基本的字符串操作并计算相关指标。其中一个度量标准帮助我们跟踪我们通常在 DNA 片段的每个位置上看到的 A、C、G 和 T 的数量。
一旦为该数据块计算了度量标准,我们就会用 Plotly.js 交互式地绘制结果,然后继续处理文件中的下一个块。将文件分成小块处理的原因仅仅是为了改进用户体验:一次处理整个文件将花费太长的时间,因为 FASTQ 文件通常是几百 GB 的。我们发现,0.5 MB 到 1 MB 之间的块大小将使应用程序的计算更加完美,并将更快地向用户返回信息,但是这个数字大小会根据应用程序的细节和计算量的大小而变化。
我们最初的 JavaScript 实现的架构相当简单:
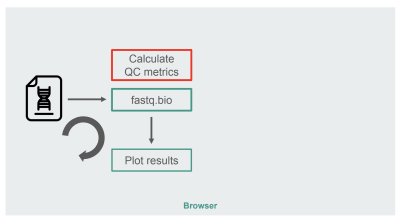
fastq.bio 的 JavaScript 实现架构。(查看大图)
红色的框是我们进行字符串操作以生成度量的地方。该框是应用程序中计算密集型的部分,这自然使它成为使用 WebAssembly 进行运行时优化的目标。
用 WebAssembly 实现 Fastq.Bio
为了探索我们是否可以利用 WebAssembly 来加速我们的 Web 应用程序,我们搜索了一个现成的工具来计算 FASTQ 文件上的 QC 指标。具体地说,我们寻找了一个用 C / C++ / Rust 编写的工具,这样它就可以移植到 WebAssembly,而且这个工具已经得到了科学界的验证和信任。
经过一些研究,我们决定使用 seqtk,这是一个用 C 语言编写的常用的开源工具,可以帮助我们评估测序数据的质量(通常用于操作这些数据文件)。
在编译到 WebAssembly 之前,让我们首先考虑如何将 seqtk 编译为二进制文件,以便在命令行上运行它。根据生成文件,你需要执行以下 gcc 命令:
# Compile to binary
$ gcc seqtk.c \
-o seqtk \
-O2 \
-lm \
-lz
另一方面,要将 seqtk 编译到 WebAssembly,我们可以使用 Emscripten toolchain,它为现有的构建工具提供了替换,从而使得在 WebAssembly 中工作更加容易。如果你没有安装 Emscripten,你可以下载我们在 Dockerhub 上准备的 docker 镜像,里面有你需要的工具(你也可以从头开始安装,但通常需要一段时间):
$ docker pull robertaboukhalil/emsdk:1.38.26
$ docker run -dt --name wasm-seqtk robertaboukhalil/emsdk:1.38.26
在容器内部,我们可以使用 emcc 编译器代替 gcc:
# Compile to WebAssembly
$ emcc seqtk.c \
-o seqtk.js \
-O2 \
-lm \
-s USE_ZLIB=1 \
-s FORCE_FILESYSTEM=1
如你所见,编译为二进制和 WebAssembly 相比差异是非常小的:
- 我们要求 Emscripten 生成一个
.wasm和.js来处理 WebAssembly 模块的实例化,而不是输出二进制文件seqtk - 为了支持 zlib 库,我们使用
USE_ZLIB标志;zlib 非常常见,它已经被移植到 WebAssembly,Emscripten 将在我们的项目中包含它 - 我们启用了 Emscripten 的虚拟文件系统,这是一个类似 POSIX 的文件系统(这里是源码),但是它在浏览器的 RAM 中运行,当你刷新页面时就会消失(除非你使用 IndexedDB 将其状态保存在浏览器中,但这应该放在另一篇文章中讨论)。
为什么是虚拟文件系统?为了回答这个问题,让我们比较一下如何在命令行上调用 seqtk 和使用 JavaScript 调用编译后的 WebAssembly 模块:
# On the command line
$ ./seqtk fqchk data.fastq
# In the browser console
> Module.callMain(["fqchk", "data.fastq"])
访问虚拟文件系统非常强大,因为这意味着我们不必重写 seqtk 来处理字符串输入。我们可以在虚拟文件系统上挂载一组数据作为文件 data.fastq,并简单地调用 seqtk 的 main() 函数。
将 seqtk 编译到 WebAssembly 后,下面是新的 fastq.bio:
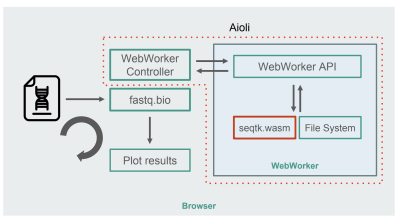
WebAssembly + WebWorkers 实现的 fastq.bio(查看大图)
如图所示,我们没有在浏览器的主线程中运行计算,而是使用 WebWorkers,它允许我们在后台线程中运行计算,从而避免对浏览器的响应性产生负面影响。具体来说,WebWorker 控制器启动 Worker 并管理与主线程的通信。在 Wroker 的一端,使用相关 API 执行它接收到的请求。
然后,我们可以要求 Worker 对刚刚挂载的文件运行 seqtk 命令。当 seqtk 运行完毕时,Worker 通过一个 Promise 将结果发送回主线程。一旦接收到消息,主线程就将结果输出来更新图表。与 JavaScript 版本类似,我们以块的形式处理文件,并在每次迭代中更新可视化视图。
优化
为了评估使用 WebAssembly 是否有任何好处,我们使用每秒可以处理多少读取数据来比较 JavaScript 和 WebAssembly。我们忽略了生成交互式图形所花费的时间,因为这两种实现方法都使用 JavaScript 来实现此目的。
开箱即见,有大约 9 倍的加速:
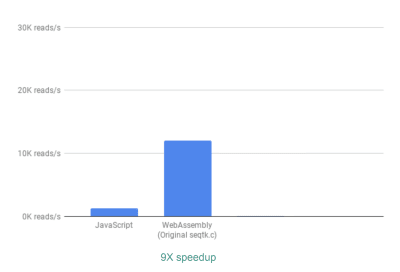
使用 WebAssembly,我们可以看到与最初的 JavaScript 实现相比,速度提高了 9 倍。(查看大图)
这已经非常好了,因为它相对来说比较容易实现(只需要你理解了 WebAssembly 即可)。
接下来,我们注意到,虽然 seqtk 输出了很多通常有用的 QC 指标,但我们的应用程序实际上并没有使用或绘制这些指标。通过删除一些我们不需要的指标的输出,可以看到一个更大的速度,13 倍:
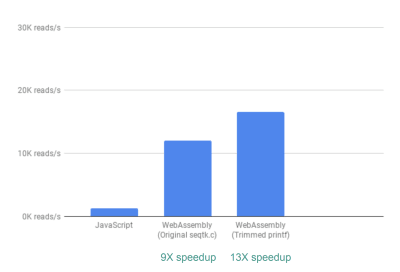
删除不必要的输出可以进一步提高性能。(查看大图)
这又是一个很大的改进,因为很容易实现,通过注释掉不需要的输出语句。
最后,我们研究了另一个改进。到目前为止,fastq.bio 获得相关度量的方法是通过两个不同的 C 函数,每个函数计算一组不同的度量。具体来说,一个函数以直方图的形式返回信息(即我们放入范围的值列表),而另一个函数以 DNA 序列位置的函数返回信息。不幸的是,这意味着相同的文件块被读取两次,这是不必要的。
因此,我们将这两个函数的代码合并到一个函数中(尽管有些混乱)。由于这两个输出的列数不同,所以我们在 JavaScript 端进行了一些区分,以便将它们分开。但这是值得的:这样做让我们实现了大于 20 倍的加速!
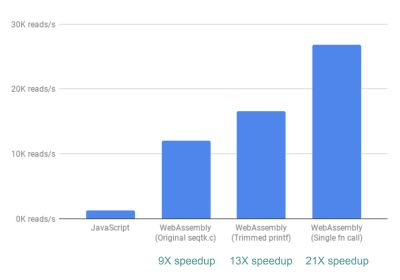
最后,使代码只读取每个文件块一次可以提高大于 20 倍的性能。(查看大图)
注意
现在是提出警告的好时机。当你使用 WebAssembly 时,不要总是期望得到 20 倍的加速。你可能只得到 2 倍的加速或者 20% 的加速。如果在内存中加载非常大的文件,或者需要在 WebAssembly 和 JavaScript 之间进行大量通信,那么速度可能会变慢。
总结
简而言之,我们已经看到用调用编译后的 WebAssembly 来替代缓慢的 JavaScript 程序可以显著提高速度。由于这些计算所需的代码已经用 C 语言实现过了,所以我们获得了重用可信工具的额外好处。正如我们还提到的, WebAssembly 并不总是适合这项工作的工具,所以要明智地使用它。
延伸阅读
- “Level Up With WebAssembly,” Robert Aboukhalil 构建web组装应用程序的实用指南。
- Aioli (on GitHub) 构建快速基因组网络工具的框架。
- fastq.bio source code (on GitHub) 一个用于 DNA 测序数据质量控制的交互式 Web 工具。
- “An Abridged Cartoon Introduction To WebAssembly,” Lin Clark
译者注:《JavaScript Weekly》周刊正在翻译中...
请戳 -> JavaScript-Weekly-zh-CN
