参考:官方文档:https:
一、GTDB-Tk安装
1.1 GTDB-Tk简介
GTDB-Tk是一个软件工具包,用于根据基因组数据库分类法(GTDB)对细菌和古细菌基因组进行客观分类。
1.2 GTDB-Tk安装
# 创建conda虚拟环境
conda create -y -n gtdbtk
# 激活环境
conda activate gtdbtk
# 安装gtdb-tk
conda install -c bioconda gtdbtk
1.3 环境检查
# gtdbtk版本检查
gtdbtk -v # version 2.3.2
1.4 下载对应数据库
# R214-最新 77.6G
wget https:
1.5 安装数据库
# 解压数据库
tar -zxvf gtdbtk_data.tar.gz -C ~/database/gtdb
# 配置GTDB数据库位置
conda env config vars set GTDBTK_DATA_PATH="~/database/gtdb"
1.6 gtdb数据库概览
ChatGPT3.5给出的结果,不一定准确
fastani
Fast Average Nucleotide Identity(FastANI)计算的结果。
FastANI用于比较两个细菌或古细菌基因组的相似性,并可用于分类和鉴定。
markers
包含一组用于分类和识别细菌和古细菌的标记基因序列。
这些标记基因通常用于构建系统发育树以确定不同基因组之间的亲缘关系。
mash
可能是与Mash算法相关的数据;
Mash用于快速比较基因组序列之间的相似性,通常用于分类和识别目的。
masks
可能是一些用于掩盖或过滤基因组序列的数据;
以便在分析过程中排除不需要的信息。
metadata
包含有关不同基因组的元数据,如来源、物种、分类信息等。
mrca_red
MRCA代表Most Recent Common Ancestor(最近的共同祖先);
这可能是用于存储最近的共同祖先数据的目录。
msa
包含多序列比对(MSA)的数据;
用于将不同基因组的序列进行对齐,以便进一步的系统发育分析。
pplacer
可能与pplacer软件相关,pplacer用于将DNA序列与系统发育树进行比对和分类。
radii
可能是与辐射分类(radii classification)相关的数据;
辐射分类通常用于分类大规模的基因组数据。
split
可能包含有关分类或分组的数据,将基因组划分为不同的类别或分类单元。
taxonomy
包含细菌和古细菌分类的数据;
例如不同分类单元的信息,包括门、纲、目、科、属和种等。
temp
临时文件或目录,用于存储临时数据。
二、GTDB-Tk用法
2.1 Check Install 检查安装
说明
check_install命令用于验证 GTDB-Tk 参考数据的完整性。
基本语法
gtdbtk check_install [--db_version DB_VERSION] [--debug] [-h]
参数解释
均为可选参数:
--db_version DB_VERSION 用于指定 GTDB-Tk 使用的数据库版本
--debug 调试模式运行
-h 帮助信息
运行gtdbtk check_install 检查
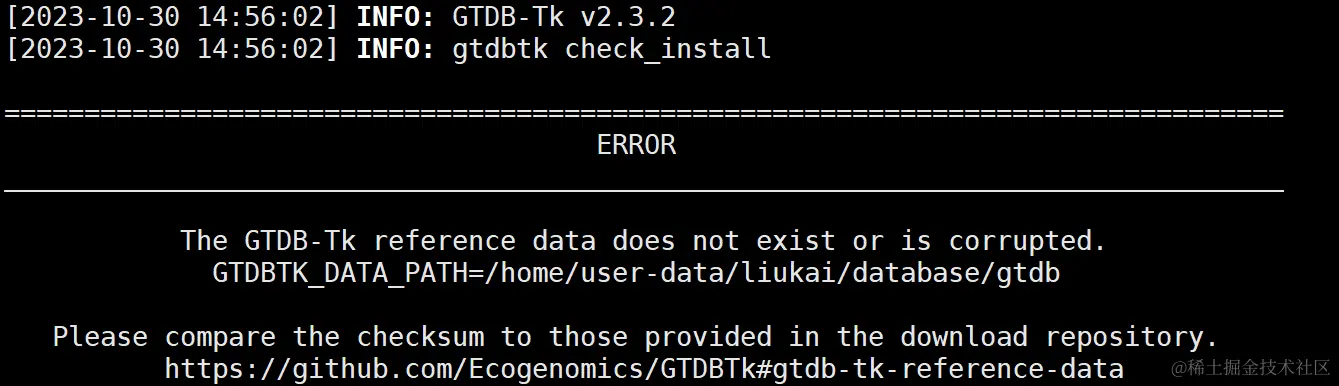
报错分析
根据报错信息,大致有三种可能造成该错误:
1、数据库文件损坏
2、安装了不兼容的数据库版本和gtdbtk工具
3、数据库路径设置错误
错误排查1--文件损坏
针对如上三个错误可能性,现逐一进行排查:
使用md5sum gtdbtk_r214_data.tar.gz 未解压的数据文件的MD5编码;
会得到这样的结果:630745840850c532546996b22da14c27 gtdbtk_r214_data.tar.gz
将结果与https:
结果一致说明文件在下载或传输过程中并能有损坏或缺失等。
错误排查2--版本不兼容
在
https:
查看gtdbtk工具版本及其支持的数据库版本;
检测自己的工具和数据库版本是否匹配。
错误排查3--路径不正确
前面提到,安装完成gtdbtk工具和gtdb数据库后要指定数据库路径;
以便在使用gtdbtk工具时要使工具能够找到数据库在哪。
使用下面的命令设置数据库路径
conda env config vars set GTDBTK_DATA_PATH="~/database/gtdb"
这里有几个容易出错的地方:
1、尽量不要使用相对路径,而是使用绝对路径:如/home/tom/database/gtdb而不是~/database/gtdb
2、检查数据库目录结构;
我们指定的目录的当前目录下应该能够看到fastani、markers等数据库文件;
比如我这里gtdb目录下只有release214文件夹;
进入release214文件夹后,发现fastani、markers等文件都在这个目录下;
所以这里设置的完整路径应该是/home/tom/database/gtdb/release214
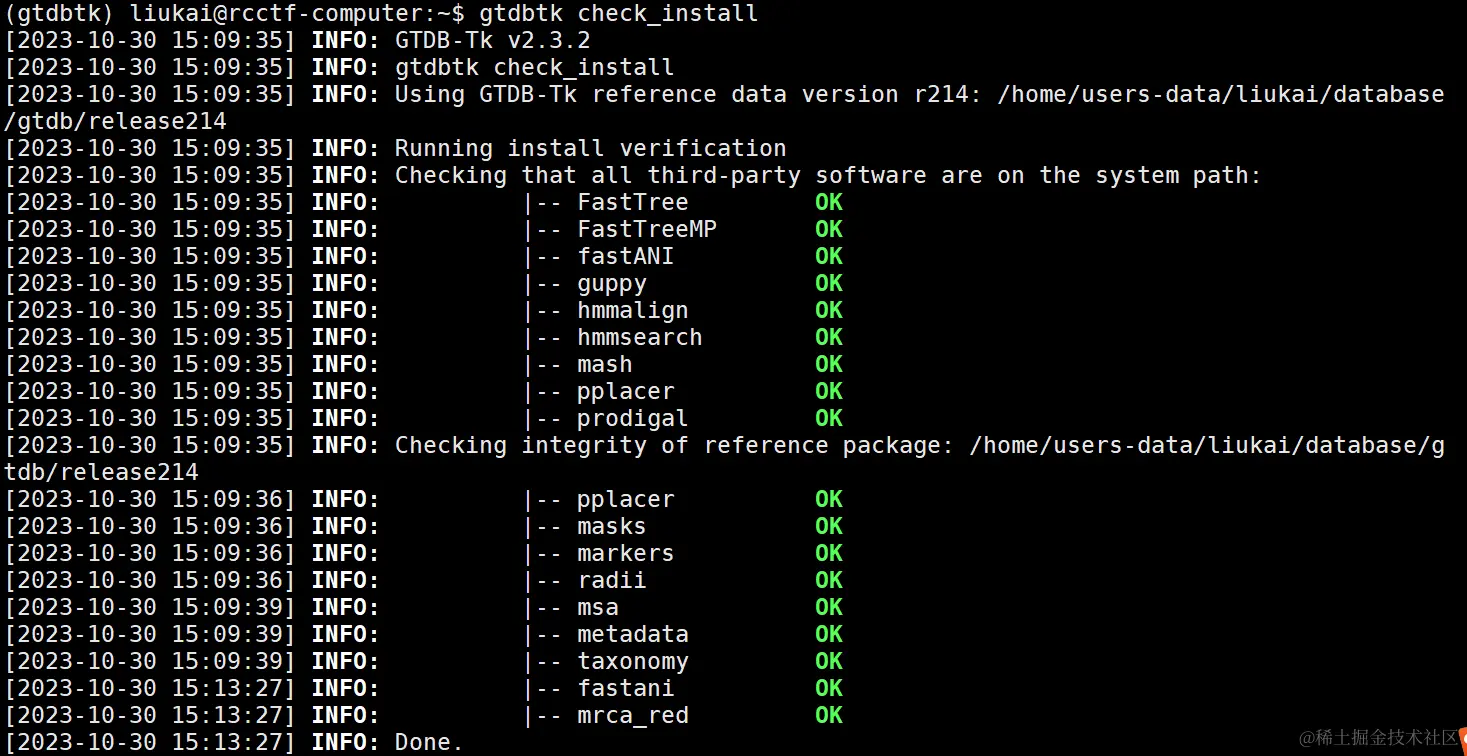
注意:重新设置数据库路径后,一定要重新启动环境:
conda deactivate # 关闭环境
conda activate gtdbtk # 重新激活环境
再次运行gtdbtk check_install 命令检查安装,输出如下结果,即安装成功
2.2 Identity 识别基因组中的标记基因
基本语法
gtdbtk identify (--genome_dir GENOME_DIR | --batchfile BATCHFILE)
--out_dir OUT_DIR [-x EXTENSION] [--prefix PREFIX]
[--genes] [--cpus CPUS] [--force]
[--write_single_copy_genes] [--tmpdir TMPDIR] [--debug]
[-h]
参数说明
# 必要参数:
--genome_dir GENOME_DIR | --batchfile BATCHFILE
指定包含要进行基因识别和分类的基因组数据的目录(GENOME_DIR)或批处理文件(BATCHFILE)
--out_dir OUT_DIR 指定输出结果的目录
# 可选参数:
-x EXTENSION 用于指定输出文件的扩展名
--prefix PREFIX 为输出文件的文件指定一个前缀
--genes 表示输入文件包含被调用的基因(跳过基因调用)
--cpus CPUS 用来指定用于分析的CPU核心数
--force 如果单个基因组发生错误,则继续处理
--write_single_copy_genes 输出未对齐的单拷贝标记基因
--tmpdir 指定临时文件目录
输出文件
[prefix].log
[prefix].json
[prefix].warning.log
identigy
[prefix].[domain].markers_summary.tsv
[prefix].translation_table_summary.tsv
[prefix].failed_fenomes.tsv
intermediate_result/marker_genes/[genome_id]/
[genome_id]_pfam_tophit.tsv
[genome_id]_pfam.tsv
[genome_id]_protein.faa
[genome_id]_protein.fna
[genome_id]_protein.gff
[genome_id]_tigrfam.out
[genome_id]_tigrfam_tophit.tsv
[genome_id]_tigrfam.tsv
prodigal_translation_table.tsv
使用示例
gtdbtk identify --genome_dir genomes/ --out_dir identify_output --cpus 3
2.3 Align 比对
说明
基于 AR53/BAC120 标记集创建一个多序列比对:
进行多序列比对,以便研究它们之间的相似性和差异性
基本语法:
gtdbtk align --identify_dir IDENTIFY_DIR --out_dir OUT_DIR
[--skip_gtdb_refs] [--taxa_filter TAXA_FILTER]
[--min_perc_aa MIN_PERC_AA]
[--cols_per_gene COLS_PER_GENE]
[--min_consensus MIN_CONSENSUS]
[--max_consensus MAX_CONSENSUS]
[--min_perc_taxa MIN_PERC_TAXA] [--rnd_seed RND_SEED]
[--prefix PREFIX] [--cpus CPUS] [--tmpdir TMPDIR]
[--debug] [-h] [--custom_msa_filters | --skip_trimming]
参数说明
# 必要参数:
--identify_dir IDENTIFY_DIR 指定了标识结果所在的目录
--out_dir OUT_DIR 指定了结果比对的输出目录
# 可选参数:
--skip_gtdb_refs 多重序列比对中不包括 GTDB 参考基因组
--taxa_filter TAXA_FILTER 将 GTDB 基因组过滤到特定分类单元(逗号分隔)
--min_perc_aa MIN_PERC_AA 指定保留基因组的最小蛋白质百分比,默认10
--cols_per_gene COLS_PER_GENE 生成 MSA 时每个基因保留的最大列数,默认42(MSA,多序列比对)
--min_consensus MIN_CONSENSUS 指定最小一致性百分比,默认25
--max_consensus MAX_CONSENSUS 指定最大一致性百分比,默认95
--min_perc_taxa MIN_PERC_TAXA 指定最小分类单元百分比,默认50
--rnd_seed RND_SEED 指定用于生成随机数的种子
--prefix PREFIX 所有输出文件的前缀
--cpus CPUS 指定使用CPU数量
--tmpdir TMPDIR 指定临时文件目录
--debug 调试模式运行,会输出更多的调试信息
-h 帮助信息
--custom_msa_filters | --skip_trimming
--custom_msa_filters
自定义过滤
`cols_per_gene`、`min_consensus` `max_consensus` `min_perc_taxa`
--skip_trimming 跳过修剪步骤返回完整的MSA
2.4 trim_msa 修剪msa
说明
修剪多序列比对 (Multiple Sequence Alignment, MSA) 数据。
对多序列比对(Multiple Sequence Alignment,MSA)数据进行修剪的目的是为了
去除不必要的信息、提高分析的准确性、减少计算负担,并简化数据的解释
基本语法
gtdbtk trim_msa --untrimmed_msa UNTRIMMED_MSA --output OUTPUT
(--mask_file MASK_FILE | --reference_mask {arc,bac})
[--debug] [-h]
参数说明
# 必要参数:
`--untrimmed_msa` 指定未修剪的多序列比对 (MSA) 文件的路径
`--output OUTPUT` 指定修剪后的 MSA 结果文件的输出路径
`--mask_file` 用于修剪 MSA 的自定义掩码文件的路径
`--reference_mask {arc,bac}` 使用参考数据库中提供的默认掩码文件来进行修剪
# 可选参数:
....
使用示例
gtdbtk trim_msa --untrimmed_msa msa.faa --output msa_trim.faa --mask_file mask.txt
2.5 export_msa
说明
将多序列比对(Multiple Sequence Alignment,MSA)的结果导出到文件或其他格式的过程。
基本语法
gtdbtk export_msa --domain {arc,bac} --output OUTPUT [--debug] [-h]
参数说明
必要参数:
--domain {arc,bac} 指定要导出 MSA 的域domain, "arc"古细菌或 "bac"细菌
--output OUTPUT 指定导出 MSA 结果的输出文件路径或目录。
可选参数:
--debug 调试模式
-h | --help 显示帮助信息
使用示例
gtdbtk export_msa --domain arc --output /tmp/msa.faa
2.6 classify
说明
确定基因组的生物分类:
"生物分类" 指的是将生物体或基因组归入不同的生物分类单元(例如,门、纲、目、科、属和种等);
以便更好地理解它们之间的"亲缘关系和系统发育关系"。
参数解释
必要参数:
--genome_dir GENOME_DIR | --batchfile BATCHFILE
选择两种方式之一,指定包含要分类的基因组数据的目录(GENOME_DIR)或批处理文件(BATCHFILE)
--align_dir ALIGN_DIR 指定包含多序列比对(MSA)结果的目录,用于分类。
--out_dir OUT_DIR 指定输出结果的目录,分类结果将保存在该目录下
--skip_ani_screen | --mash_db MASH_DB
用于指定ANI(Average Nucleotide Identity)筛选方式。
--skip_ani_screen 表示跳过ANI筛选;
--mash_db MASH_DB 表示使用Mash数据库来进行ANI筛选
# 可选参数
--no_mash 跳过使用 Mash 预过滤(pre-filtering)基因组的步骤
--mash_k MASH_K 设置Mash算法的k-mer大小,默认是16,(1-32)
--mash_s MASH_S 设置Mash算法的sketch大小,默认5000
--mash_v MASH_V 设置Mash算法的p-value的最大值,默认是1.0,(0-1)
--mash_max_distance MASH_MAX_DISTANCE 设置Mash算法的最大距离阈值,默认0.15
-x EXTENSION 指定要处理文件的扩展名,默认是fna
--prefix PREFIX 输出文件的文件名前缀,默认是gtdbtk
--cpus CPUS 指定用于计算的CPU核心数,默认是1
--pplacer_cpus PPLACER_CPUS 指定用于运行pplacer的CPU核心数
--scratch_dir SCRATCH_DIR 指定一个用于存储临时数据的目录
--genes 保留用于分类的基因标记数据
-f 覆盖已存在的输出文件
--min_af MIN_AF 设置ANI筛选的最小比对分数阈值
--tmpdir TMPDIR 指定临时文件目录,默认是/temp
--debug 在调试模式下运行,输出更多的调试信息
-h | --help 显示帮助信息
输出文件
classify
[prefix].[domain].summary.tsv
[prefix].backbone.[domain].classify.tree
[prefix].[domain].tree.mapping.tsv
[prefix].[domain].classify.tree.[index]tree
intermediate_result # 中间结果
[prefix].[domain].backbone.classification_pplacer.tsv
[prefix].[domain].class_level.classfication_pplacer_tree_[index].tsv
[prefix].[domain].prescreened.msa.fasta
[prefix].[domain].red_dictionary.tsv
ani_screen
intermediate_results
mash
[prefix].mash_distances.tsv
[prefix].user_query_sketch.msh
[prefix].[domain].summary.tsv
[prefix].log
[prefix].json
[prefix].warning.log
使用示例
gtdbtk classify --align_dir align_3lines/ --batchfile 3lines_batchfile.tsv --out_dir 3classify_ani --mash_db mash_db_dir/ --cpus 20
2.7 ani_rep
说明
计算输入基因组与所有 GTDB-Tk 代表性基因(参考基因)组的ANI。
什么是ANI:
ANI 是 Average Nucleotide Identity 的缩写;
指的是核苷酸的平均相似性。
ANI 是一种计算两个细菌或古细菌基因组之间相似性的度量方法;
通常用于确定它们之间的亲缘关系和分类。
ANI 的计算步骤:
选择要比较的两个基因组;
从这两个基因组中选择一组核苷酸序列,通常是基因组中的特定基因或标记基因;
对选定的核苷酸序列进行比对;
根据比对的结果计算平均相似性,这通常以百分比表示;
基本语法
基本语法:
gtdbtk ani_rep (--genome_dir GENOME_DIR | --batchfile BATCHFILE)
--out_dir OUT_DIR [--no_mash] [--mash_k MASH_K]
[--mash_s MASH_S] [--mash_d MASH_D] [--mash_v MASH_V]
[--mash_db MASH_DB] [--min_af MIN_AF] [-x EXTENSION]
[--prefix PREFIX] [--cpus CPUS] [--tmpdir TMPDIR]
[--debug] [-h]
参数解释
参数解释:
必要参数:
--genome_dir GENOME_DIR | --batchfile BATCHFILE
选择两种方式之一,指定包含要计算ANI的基因组数据的目录(GENOME_DIR)或批处理文件(BATCHFILE)
--out_dir OUT_DIR 指定计算结果输出的目录
可选参数:
--no_mash 跳过使用 Mash 预过滤(pre-filtering)基因组的步骤
--mash_k MASH_K 设置Mash算法的k-mer大小,默认是16,(1-32)
--mash_s MASH_S 设置Mash算法的sketch大小,默认5000
--mash_d MASH_D 设置Mash算法的距离阈值,默认是0.15,(0-1)
--mash_v MASH_V 设置Mash算法的p-value的最大值,默认是1.0,(0-1)
--mash_db MASH_DB 指定Mash数据库的位置,用于ANI计算
--min_af MIN_AF 设置ANI计算中的最小比对分数阈值,默认是0.5
-x EXTENSION 指定要处理文件的扩展名,默认是fna
--prefix PREFIX 输出文件的文件名前缀,默认是gtdbtk
--cpus CPUS 指定用于计算的CPU核心数,默认是1
--tmpdir TMPDIR 指定临时文件目录,默认是/temp
--debug 在调试模式下运行,输出更多的调试信息
-h | --help 显示帮助信息
输出文件
[prefix].ani_closest.tsv
[prefix].ani_summary.tsv
[prefix].log
[prefix].warnings.log
intermediate_results/mash/
[prefix].gtdb_ref_sketch.msh
[prefix].mash_distances.msh
[prefix].user_query_sketch.msh
使用示例
gtdbtk ani_rep --genome_dir genomes/ --out_dir ani_rep/ --cpus 70
其他-Mash
Mash 是一种用于快速估算基因组之间相似性的工具;
通常用于在大量基因组数据中找到相似的序列。
2.8 classify_wf
基本语法
gtdbtk classify_wf (--genome_dir GENOME_DIR | --batchfile BATCHFILE)
--out_dir OUT_DIR
(--skip_ani_screen | --mash_db MASH_DB) [--no_mash]
[--mash_k MASH_K] [--mash_s MASH_S]
[--mash_v MASH_V]
[--mash_max_distance MASH_MAX_DISTANCE] [-f]
[-x EXTENSION] [--min_perc_aa MIN_PERC_AA]
[--prefix PREFIX] [--genes] [--cpus CPUS]
[--pplacer_cpus PPLACER_CPUS] [--force]
[--scratch_dir SCRATCH_DIR]
[--write_single_copy_genes] [--keep_intermediates]
[--min_af MIN_AF] [--tmpdir TMPDIR] [--debug] [-h]
参数解释
必要参数:
--genome_dir GENOME_DIR | --batchfile BATCHFILE
两种方式之一,指定包含要分类的基因组数据的目录(GENOME_DIR)或批处理文件(BATCHFILE)
--out_dir OUT_DIR 指定输出结果的目录,分类结果保存在该目录下
--skip_ani_screen | --mash_db MASH_DB
用于指定ANI(Average Nucleotide Identity)筛选方式;
--skip_ani_screen 表示跳过ANI筛选,
--mash_db MASH_DB 表示使用Mash数据库来进行ANI筛选
可选参数:
--no_mash 跳过使用 Mash 预过滤(pre-filtering)基因组的步骤
--mash_k MASH_K 设置Mash算法的k-mer大小,默认是16,(1-32)
--mash_s MASH_S 设置Mash算法的sketch大小,默认5000
--mash_v MASH_V 设置Mash算法的p-value的最大值,默认是1.0,(0-1)
--mash_max_distance MASH_MAX_DISTANCE 设置Mash算法的最大距离阈值
-f 覆盖已存在的输出文件
-x EXTENSION 指定输出文件的扩展名
--min_perc_aa MIN_PERC_AA 设置用于分类的最小氨基酸百分比阈值,默认10
--prefix PREFIX 为输出文件的文件名指定一个前缀
--genes 保留用于分类的基因标记数据
--cpus CPUS 指定用于分类的CPU核心数
--pplacer_cpus PPLACER_CPUS 指定用于运行pplacer的CPU核心数
--force 如果单个基因组发生错误,则继续处理
--scratch_dir SCRATCH_DIR 通过写入磁盘(较慢)来减少 pplacer 内存使用量
--write_single_copy_genes 输出未对齐的单拷贝标记基因
--keep_intermediates 将保留中间文件
--min_af MIN_AF 设置ANI筛选的最小比对分数阈值,默认0.5
--tmpdir TMPDIR 指定临时文件目录
--debug 调试模式运行
-h | --help 显示帮助信息
使用示例
gtdbtk classify_wf --genome_dir genomes/ --out_dir classify_wf_out --cpus 3
2.9 convert_to_itol 转换为iTOL格式
说明
`convert_to_itol`
是一个用于将生物信息学数据转换为 iTOL(Interactive Tree of Life)格式的工具或脚本。
`iTOL`
是一个用于可视化和呈现生命树(Phylogenetic Tree)和分类数据的在线工具;
它允许用户创建交互式、可定制的生命树图表。
基本语法
gtdbtk convert_to_itol --input_tree INPUT_TREE
--output_tree OUTPUT_TREE
[--debug]
[-h]
参数解释
--input_tree INPUT_TREE 指定输入的 Newick 树(生命树)文件的路径
--output_tree OUTPUT_TREE 指定输出文件路径
使用示例
gtdbtk convert_to_itol --input some_tree.tree --output itol.tree
2.10 de_novo_wf 从头开始工作流
说明
基本语法
gtdbtk de_novo_wf (--genome_dir GENOME_DIR | --batchfile BATCHFILE)
(--bacteria | --archaea) --outgroup_taxon
OUTGROUP_TAXON --out_dir OUT_DIR [-x EXTENSION]
[--skip_gtdb_refs] [--taxa_filter TAXA_FILTER]
[--min_perc_aa MIN_PERC_AA] [--custom_msa_filters]
[--cols_per_gene COLS_PER_GENE]
[--min_consensus MIN_CONSENSUS]
[--max_consensus MAX_CONSENSUS]
[--min_perc_taxa MIN_PERC_TAXA] [--rnd_seed RND_SEED]
[--prot_model {JTT,WAG,LG}] [--no_support] [--gamma]
[--gtdbtk_classification_file GTDBTK_CLASSIFICATION_FILE]
[--custom_taxonomy_file CUSTOM_TAXONOMY_FILE]
[--write_single_copy_genes] [--prefix PREFIX]
[--genes] [--cpus CPUS] [--force] [--tmpdir TMPDIR]
[--keep_intermediates] [--debug] [-h]
参数解释
必要参数:
--genome_dir GENOME_DIR | --batchfile BATCHFILE
指定包含要进行系统发育分析的基因组数据的目录(GENOME_DIR)或批处理文件(BATCHFILE)
--bacteria | --archaea
用于指定要分析的基因组是细菌还是古细菌
--outgroup_taxon OUTGROUP_TAXON 指定一个外类群(outgroup)用于进行根化(rooting)系统发育树,以确定根的位置。
--out_dir OUT_DIR 指定输出结果的目录
可选参数:
-x EXTENSION 可以用来指定输出文件的扩展名
--skip_gtdb_refs 多重序列比对中不包括 GTDB 参考基因组
--taxa_filter 将 GTDB 基因组过滤到特定分类组内的分类单元
--min_perc_aa MIN_PERC_AA 设置最小的氨基酸百分比阈值
--custom_msa_filters 使用自定义多序列比对过滤器
--cols_per_gene 指定生成多序列比对时每个基因保留的最大列数
--min_consensus 设置一致性过滤的最小阈值
--max_consensus 设置一致性过滤的最大阈值。
--min_perc_taxa 设置最小百分比的分类单元
--rnd_seed 设置随机种子
--prot_model {JTT,WAG,LG} 用来选择蛋白质进化模型,用于系统发育分析
--no_support 不使用 Shimodaira-Hasekawa 检验计算局部支持值
--gamma 进行分支长度的重新缩放,以优化使用 Gamma 分布模型计算的似然度
--gtdbtk_classification_file 指定 GTDB-Tk 分类文件的路径
--custom_taxonomy_file 指定自定义的分类文件的路径
--write_single_copy_genes 输出未对齐的单拷贝标记基因
--prefix PREFIX 为输出文件的文件指定一个前缀
--genes 执行基因级别的分析
--cpus CPUS 指定用于分析的CPU核心数
--force 如果单个基因组发生错误,则继续处理
--tmpdir 指定临时文件目录
--keep_intermediates 保留中间文件
--debug 调试模式
-h | --help 显示帮助信息
使用示例
gtdbtk de_novo_wf --genome_dir genomes/ --outgroup_taxon p__Undinarchaeota --archaea --out_dir de_novo_wf --cpus 3
gtdbtk de_novo_wf --genome_dir genomes/ --outgroup_taxon p__Chloroflexota --bacteria --taxa_filter p__Firmicutes --out_dir de_novo_output
2.11 infer 推断
说明
用于执行系统发育推断
基本语法
gtdbtk infer --msa_file MSA_FILE --out_dir OUT_DIR
[--prot_model {JTT,WAG,LG}] [--no_support] [--gamma]
[--prefix PREFIX] [--cpus CPUS] [--tmpdir TMPDIR]
[--debug] [-h]
参数说明
# 必要参数:
--msa_file MSA_FILE 指定多序列比对(MSA)文件(FASTA)的路径,该文件将用于进行系统发育推断
--out_dir OUT_DIR 指定输出结果的目录
# 可选参数:
--prot_model {JTT,WAG,LG} 选择蛋白质模型,以用于系统发育分析,默认WAG
--no_support 不使用 Shimodaira-Hasekawa 检验计算局部支持值
--gamma 重新调整分支长度以优化 Gamma20 似然
--prefix PREFIX 为输出文件的文件指定一个前缀
--cpus CPUS 指定用于系统发育推断的CPU核心数
--tmpdir TMPDIR 指定临时文件目录
输出文件
[prefix].log
[prefix].unrooted.tree
[profix].warning.log
inter/intermediate_results/
[prefix].fasttree.log
[prefix].tree.log
使用示例
gtdbtk infer --msa_file msa.faa --out_dir infer_out
2.12 Root 根化
说明
用于对系统发育树进行`根化(rooting)`。
`根化`是将系统发育树的根节点(root node)放置在所选的外类群(outgroup)物种或分类单元上;
以更好地理解和解释树的拓扑结构和进化关系。
基本语法
gtdbtk root --input_tree INPUT_TREE --outgroup_taxon OUTGROUP_TAXON
--output_tree OUTPUT_TREE
[--gtdbtk_classification_file GTDBTK_CLASSIFICATION_FILE]
[--custom_taxonomy_file CUSTOM_TAXONOMY_FILE]
[--tmpdir TMPDIR] [--debug] [-h]
参数说明
# 必要参数:
--input_tree 指定输入的系统发育树文件的路径,即需要进行根化的树
--outgroup_taxon 指定外类群(outgroup)物种或分类单元的名称或标识,将使用这个外类群来根化树,例如(`p__Patescibacteria`或`p__Altiarchaeota`)。
--output_tree 指定输出的系统发育树文件的路径
# 可选参数:
--gtdbtk_classification_file 指定 GTDB-Tk 分类文件的路径,用于提供额外的分类信息`--tmpdir TMPDIR`:可选项,用来指定临时文件目录
--custom_taxonomy_file 指定自定义的分类文件的路径,用于提供树的自定义分类信息
--tmpdir TMPDIR 用来指定临时文件目录
使用示例
gtdbtk root --input_tree input.tree --outgroup_taxon p__Nanoarchaeota --output_tree output.tree
2.13 decorate 装饰
说明
"decorate" 意味着对树进行美化和装饰,以增强可视化效果并传达更多的信息。
基本语法
gtdbtk decorate --input_tree INPUT_TREE --output_tree OUTPUT_TREE
[--gtdbtk_classification_file GTDBTK_CLASSIFICATION_FILE]
[--custom_taxonomy_file CUSTOM_TAXONOMY_FILE]
[--tmpdir TMPDIR] [--debug] [-h]
参数说明
必要参数:
--input_tree INPUT_TREE 指定希望进行装饰的树的路径
--output_tree OUTPUT_TREE 指定输出的系统发育树文件的路径
可选参数:
--gtdbtk_classification_file 指定 GTDB-Tk 分类文件的路径,用于提供额外的分类信息
--custom_taxonomy_file 指定自定义的分类文件的路径
--tmpdir TMPDIR 指定临时文件目录
使用示例
gtdbtk decorate --input_tree input.tree --output_tree output.tree
2.14 test 测试
说明
`test`命令用于通过分类工作流程运行三个小型古菌基因组。
基本语法
gtdbtk root --input_tree INPUT_TREE --outgroup_taxon OUTGROUP_TAXON
--output_tree OUTPUT_TREE
[--gtdbtk_classification_file GTDBTK_CLASSIFICATION_FILE]
[--custom_taxonomy_file CUSTOM_TAXONOMY_FILE]
[--tmpdir TMPDIR] [--debug] [-h]
参数说明
# 必要参数:
`--out_dir OUT_DIR` 指定输出测试结果的目录
# 可选参数:
`--tmpdir TMPDIR` 用来指定临时文件目录
使用示例
gtdbtk test --out_dir /tmp/test --cpus 3
三、GTDBTk工作流
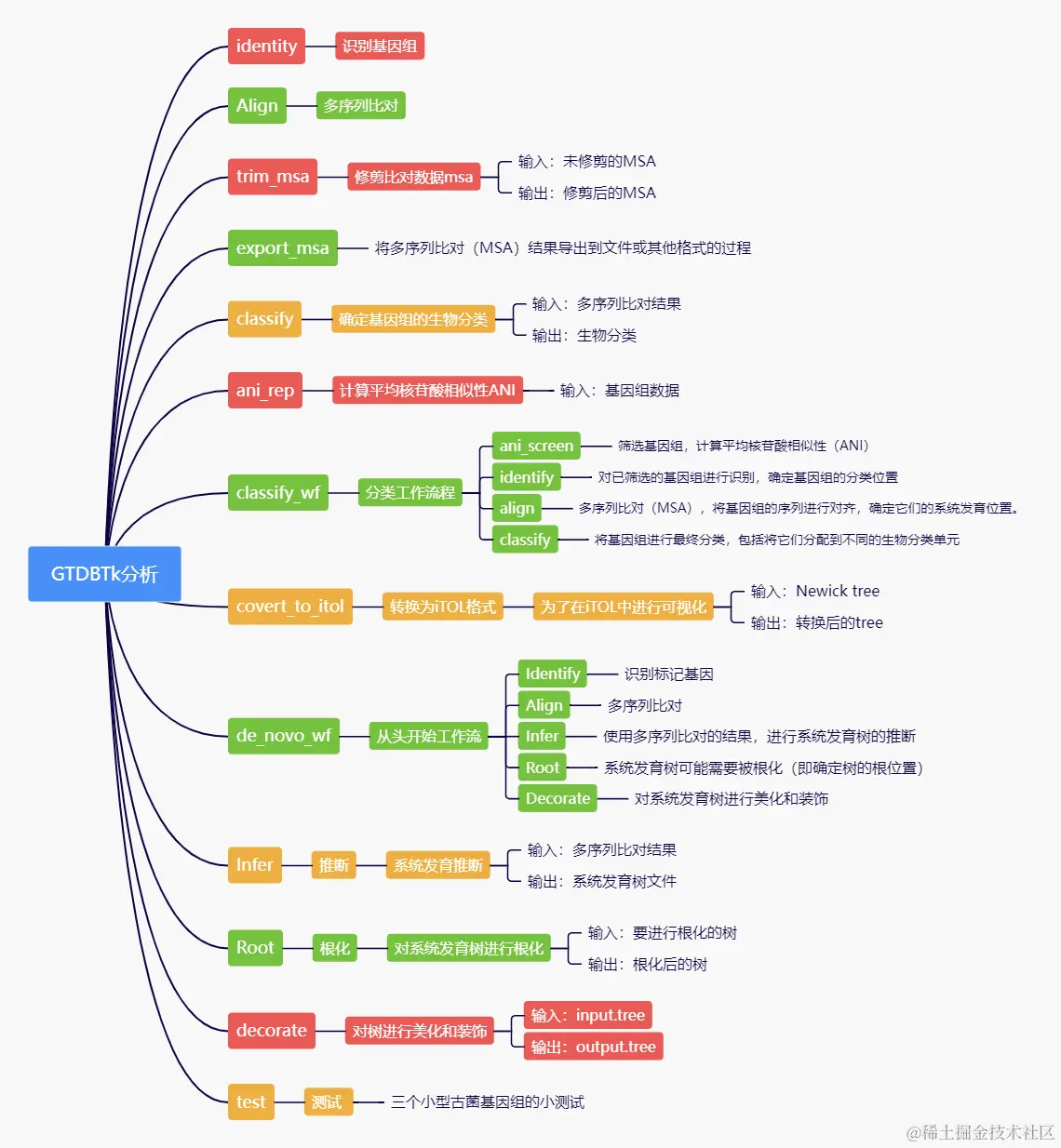
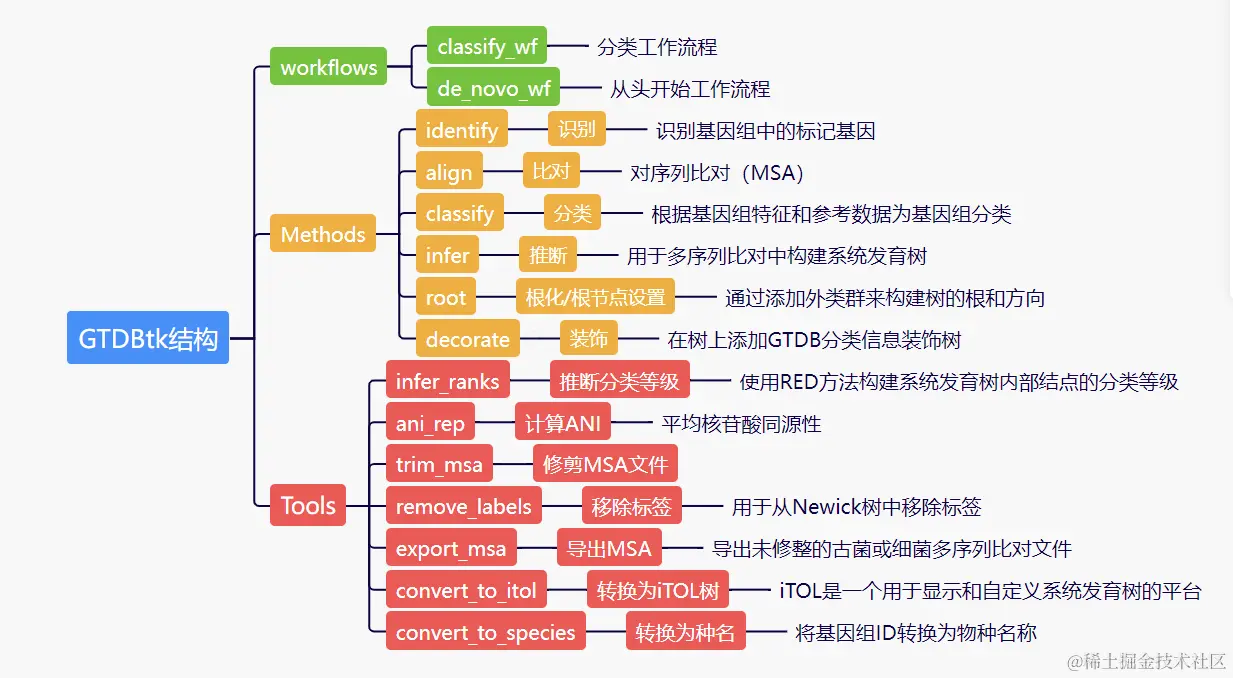
四、GTDBtk完整结构
五、使用演示
......


